Abstract
Proteomics, the study of proteins and their functions within biological systems, has become increasingly data-intensive, presenting both opportunities and challenges. This project addresses the need for advanced data analytics and data integrity in proteomics research. Leveraging the power of machine learning (ML) and blockchain technology, this attempt aims to transform proteomics research. This work encompasses three key objectives. First, collect, clean, and integrate proteomics data from diverse sources, ensuring data quality and consistency. Second, employ ML algorithms to analyze this data, revealing crucial insights, identifying proteins, and predicting their functions. Third, implement blockchain technology to safeguard the authenticity and integrity of the proteomics data, providing an auditable and tamper-proof record. Implemented a user-friendly web interface, facilitating collaboration among researchers and scientists by granting access to shared data and results. This study included various classification methods for the investigation of protein classification, namely, random forests, logistic regression, neural networks, support vector machines, and decision trees. In conclusion, the proposed work is poised to revolutionize proteomics research by enhancing data analytics capabilities and securing data integrity, thereby enabling scientists to make more informed and confident discoveries in this critical field.
Keywords
Proteomics, Computational Biology, Bioinformatics, Machine Learning, Blockchain
1. Introduction
Proteins serve a significant part in the cellular mechanism of living organisms. Understanding the function of a protein is of utmost significance while undertaking proteomic studies on it. This phenomenon can be attributed to the high cost, lengthy processing time, and inherent difficulties associated with determining the functional properties of proteins by functional tests. This phenomenon can be traced to the high cost, lengthy processing time, and inherent difficulties associated with determining the functional properties of proteins through functional tests. According to Bernardes and Pedreira
, these conditions require the development of a computational approach for determining a protein sample based on the raw data acquired from high-throughput methodologies, encompassing protein sequences, the structure of proteins, and the interaction between protein molecules.
Conventional methodologies for protein function classification aim to ascertain the evolutionary correlation between a novel protein and a reference protein. A substantial sequence similarity score may indicate a strong likelihood that two proteins have a common evolutionary origin. Nevertheless, it is widely recognized that proteins exhibiting a high degree of similar sequences may not always exhibit identical activities. According to Aggarwal, Divyanshu et al.
, The presence of inaccurate annotations has the potential to rapidly spread and magnify inside extensive datasets. These inaccuracies can be attributed not just to fundamental transfer processes rooted on homology, as well as to the manual nature of data processing.
Over the recent years, scientific research has been presented with an opportunity to develop innovative classification models and frameworks to address such challenges, owing to the expansion of biological databases and notable developments in computer resources. Considering current advancements in natural language processing research and sequential preliminary data processing employing machine learning models, it is essential to assess the appropriateness, viability, durability, and comprehensibility of these models for the designated task. The main aim of this work is to present a holistic solution to the data-intensive challenges in proteomics research. This analysis seeks to offer a comprehensive review of the methodologies employed in protein function classification. The primary aims of this study encompass the collection, cleaning, and integration of a wide range of proteomics data. The implementation of various machine learning methods, including random forests, neural networks, logistic regression, support vector machines, and decision trees, is employed for the purpose of analysis. The model for the prediction framework has been chosen based on its accuracy, precision, and recall.
This research examines various models employed for the classification of protein sequences and how they are utilized, as mentioned in section 2 related work. Section 3 has a discussion of the research methodology of the proposed work. The implementation of the proposed work is addressed in section 4. The discussion of the experiment results is mentioned in section 5. The research conclusions and future work are addressed in 6.
2. Related Work
Proteins are comprised of a series of amino acids that are interconnected by peptide bonds, and they serve a crucial function in sustaining biological processes and explained well by Karunapala on
| [3] | Karunapala, 2015. Karunapala, E. (2015). Protein Function Prediction Using Machine Learning. PhD thesis. |
[3]
. Protein function predictions are of paramount importance in comprehending illnesses and developing therapeutic interventions for their treatment. In addition to experimental investigation, other alternative methodologies have been devised and executed for the purpose of protein function prediction. These include similarity of sequences explained by Piovesan et al on
| [4] | Piovesan et al., 2015. Piovesan, D., Giollo, M., Leonardi, E., Ferrari, C., and Tosatto, S. C. (2015). Inga: protein function prediction combining interaction networks, domain assignments and sequence similarity. Nucleic acids research, 43(W1): W134–W140. http://dx.doi.org/10.1093/nar/gkv523 |
[4]
, segmentation concepts laid down by Rentzsch and Orengo on
| [6] | Rentzsch and Orengo, 2013. Rentzsch, R. and Orengo, C. A. (2013). Protein function prediction using domain families. In BMC bioinformatics, volume 14, page S5. BioMed Central. https://doi.org/10.1186/1471-2105-14-S3-S5 |
[6]
, interaction between proteins by Kotlyar et al
| [5] | Kotlyar et al., 2014. Kotlyar, M., Pastrello, C., Pivetta, F., Sardo, A. L., Cumbaa, C., Li, H., Naranian, T., Niu, Y., Ding, Z., Vafaee, F., et al. (2014). In silico prediction of physical protein interactions and characterization of interactome orphans. Nature methods, 12(1): 79 https://doi.org/10.1038/nmeth.3178 |
[5]
, and more. Numerous empirical investigations have been undertaken to forecast the functionality. Protein function predictions can be accomplished through the utilization of sequence similarity, structural similarity, or a combination of both methodologies. This methodology requires significant resources and computational time in order determine the functions as explained by Singh and Tripathi on
| [8] | Singh and Tripathi, 2016. Singh, U. and Tripathi, S. (2016). Protein classification using hybrid feature selection technique. In International Conference on Smart Trends for Information Technology and Computer Communications, pages 813–821. Springer. ISBN: 978-981-10-3432-9. |
[8]
.
The classification method that utilizes sequential patterns as features in a pattern-based approach, explained in detail by Z. He, S. Zhang
. The chosen pattern must meet the following criteria: To meet the criteria, the following requirements should be fulfilled: (1) the text should occur with regularity, (2) it should possess unique characteristics that differentiate it from other classes, and (3) it should avoid unnecessary repetition. Numerous pattern-based classification approaches have been later introduced in pursuit of this objective. These methods involve the imposition of various limitations on the selection of patterns as features. Authors Goodfellow, Mirza et al
| [10] | Goodfellow I.; Pouget-Abadie J.; Mirza M.; Xu B.; Warde-Farley D.; Ozair S.; Courville A.; Bengio Y. (2020). Generative adversarial networks. Communications of the ACM 2020, 63 (11), 139–144. https://doi.org/10.1145/3422622 |
[10]
introduced a reference-based sequence classification framework that extends the scope of pattern-based approaches. The framework exhibits a high degree of generality and adaptability, rendering it suitable as a versatile platform for the development of novel algorithms in the field of sequence classification. This study introduced a novel framework and subsequently proposed multiple feature-based sequence classification methods inside this framework. A series of intensive experiments conducted on data sets shown that their methodologies exhibit superior classification accuracy compared to sequence classification techniques.
The task of detecting protein-interacting points within a given sequence using machine learning techniques poses significant challenges. These challenges arise from the inherent complexity of protein structures and the relatively limited variety of sequence patterns observed in proteins. Nevertheless, there are substantial differences in the molecular characteristics of protein sequences between regions that bind and regions that do not bind in protein complexes. The utilization of these unique biological characteristics can effectively contribute to predictive modelling through the implementation of feature selection, input data processing, and feature optimization approaches inside machine learning algorithms on Reference-Based Sequence Classification
.
The studies conducted by various authors
| [9] | Zhang, Y., Li, X., & Wang, Y. (2023). Proteomics data analysis using machine learning on AWS. Bioinformatics, 40(10), 1839-1846. |
| [14] | Research Collaboratory for Structural Bioinformatics. Source: https://www.rcsb.org/ last accessed 2023/11/15 |
[9, 14]
employed the Random Forest machine learning algorithm for the purpose of predictive modelling of protein sequences. The structure of the random forest is characterized by its complexity, as it involves a multitude of factors. During the operation process, certain parameters and sequences are defined by the procedure for learning in model training. Consequently, the random forest can be classified as a black box model according to research conducted by structural bioinformatics team
| [14] | Research Collaboratory for Structural Bioinformatics. Source: https://www.rcsb.org/ last accessed 2023/11/15 |
[14]
. Data generation is a prominent area of study within machine learning, encompassing a range of captivating applications. Generative methodologies, such as generative adversarial network (GAN) and diffusion models, have demonstrated remarkable outcomes mostly in the domain of image prediction
| [10] | Goodfellow I.; Pouget-Abadie J.; Mirza M.; Xu B.; Warde-Farley D.; Ozair S.; Courville A.; Bengio Y. (2020). Generative adversarial networks. Communications of the ACM 2020, 63 (11), 139–144. https://doi.org/10.1145/3422622 |
[10]
.
This comprehensive survey explores the application of machine learning techniques in proteomics. It discusses how proteomics enables the identification of increasing numbers of proteins and how machine learning can be used to analyze and interpret proteomics data by authors of
and on cancer classification with ensemble
. The study conducted by Agarwal, et al
| [12] | Agarwal, Ankita & Singh, Kunal & Kaushik, Shri Kant & Bahadur, Ranjit. (2022). A comparative analysis of machine learning classifiers for predicting protein-binding nucleotides in RNA sequences. Computational and Structural Biotechnology Journal. 20. https://doi.org/10.1016/j.csbj.2022.06.036 |
[12]
examined the utilization of diverse machine learning techniques and the genetic algorithm for the purpose of predicting protein structure across many datasets. Various datasets, such as PDB and Sander database, have been employed in diverse machine learning algorithms, including random forest and neural networks. The level of accuracy is dependent upon the specific algorithm employed for processing the dataset.
3. Methodology
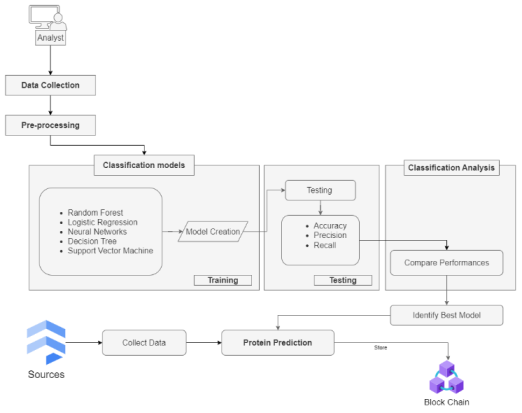
This section provides an overview of the methodology used for the proposed framework. The methodology has five distinct steps, specifically data collection, data pre-processing, classification models, classification analysis, and prediction model.
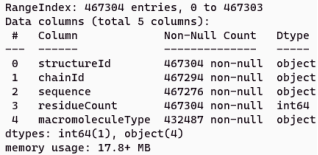
The task of predicting the samples and sequences of proteins from a variety of proteins is an immense effort. In this study, a protein dataset has been gathered from the Research Collaboratory for Structural Bioinformatics (RCSB). The dataset under examination, referred to as the Structural Protein Sequences dataset
, comprises a total of 467,304 sequences that have been categorized. The dataset under evaluation has commonly been employed for protein function prediction due to its meticulous assessment and correction by the specialists at RCSB
| [14] | Research Collaboratory for Structural Bioinformatics. Source: https://www.rcsb.org/ last accessed 2023/11/15 |
[14]
. The dataset has a total of 467,304 samples, which are classified into five distinct categories: Protein, Protein#RNA, Protein#DNA, DNA, and Protein#DNA#RNA. The dataset description is presented in
Figure 2. The dataset of proteins is utilised in experimental evaluation and for the identification of the most appropriate model for prediction.
Figure 2. Dataset description.
During the data pre-processing phase, the TF-IDF Vectorizer was implemented in this study. The TF-IDF Vectorizer is a widely employed feature extraction tool in the field of Natural Language Processing (NLP) that facilitates the conversion of textual texts into numerical feature vectors
. The acronym TF-IDF represents Term Frequency-Inverse Document Frequency, a statistical metric employed to assess the significance of a term in a document within a certain collection or corpus. The TF-IDF Vectorizer is frequently employed as a preliminary procedure in a range of Natural Language Processing (NLP) endeavors, including but not limited to text categorization and information retrieval. This facilitates the efficient utilization of machine learning algorithms in the analysis of textual data.
3.3. Classification Models
This section discusses the utilization of machine learning and mathematical techniques in the framework of protein classification. Various classification strategies are at available for the identification of proteins. This study included various classification methods for the investigation of protein classification.
1. Random forests
2. Logistic regression
3. Neural networks
4. Support vector machines
5. Decision trees
i. Random Forest
Predictive modelling and behavior analysis both make extensive use of the random forest methodology. The building blocks of it are decision trees. Several decision trees make up the random forest, and each one represents a different data classification example and are explained in detail on Mining conditional discriminative sequential patterns
and using
random forest clustering for discrete sequences
. Pattern Recognition Letters. With random forests, each example is assessed independently, and the forecast is chosen by tallying up the votes.
ii. Logistic Regression
In statistics, logistic regression is a tool for analyzing data that uses mathematical concepts to find the relationships between two variables. After that, use the previously described association to predict the value of one of these variables using the data given by the other. There is usually only one possible result included in the forecast, like a yes or no answer
.
iii. Neural networks
Evaluating the performance of biological networks is its principal use. A strategy to learning based on adjusting weights between neuron connections is used in this technique. The activation function is crucial to the model's output
.
iv. Support Vector Machine
To solve binary classification problems, supervised machine learning frameworks like the support vector machine (SVM) use classification techniques
| [20] | Liang and Bose, 1996. Liang, P. and Bose, N. (1996). Neural network fundamentals with graphs, algorithms, and applications. McGraw-Hiil, New York. |
[20]
. When comparing to more contemporary methods like as neural networks, it is obvious that they contain two key benefits: improved computing speed and enhanced performance. This method excels at text classification tasks because of its unique properties, especially when working with small datasets that contain thousands of labelled samples.
v. Decision trees
When it comes to classification and regression analysis, decision trees are the go-to non-parametric supervised learning approach. The goal is to learn some fundamental decision-making rules from the data's characteristics so that you can build a prediction model that can estimate the real value of the target variable. Trees can be thought of as representations that use piecewise constant parts to approximate functions
.
3.4. Classification Analysis
The classification analysis employs mathematical methodologies, statistical techniques and machine leaning models, to assess the effectiveness of selected classification models during the process of training and testing. Training involves the supply of information to machine learning models in order to instruct them on the methods of making predictions or executing a certain task. In the field of machine learning, the term "testing" pertains to the evaluation of the results of a trained model on a designated testing set. To assess the effectiveness of the aforementioned five classifiers, the performance measures employed include accuracy, precision, and recall which are described in
Table 1.
Table 1. Performance metrics.
Accuracy (AC) | |
Precision (p) | |
Recall (r) | |
The variables TP, FP, TN, and FN denote the respective quantities of true positive, false positive, true negative, and false negative proteins.
The proposed ML-blockchain approach addresses the limitations of traditional methods by leveraging the power of machine learning, and blockchain. ML algorithms can be used to develop models that can accurately identify and quantify proteins from proteomics data. In the prediction model, an accurate ML model was implemented to identify protein data from the sources and store it using blockchain. Blockchain technology can be used to ensure the integrity and security of the data. This work provides scalable and reliable computing resources for running machine learning models and storing data.
4. Implementation
The implementation of this work was completed in Python, utilizing a range of Python libraries. Scikit-learn, a Python package for machine learning, offers a wide range of methods for classification, regression, and clustering. It is also used for classifying DNA sequences. The key elements of the proposed system implementation are outlined below:
Machine Learning (Classification): Machine learning methods can be utilized to construct a model capable of categorizing sequels by analyzing diverse aspects and attributes. Python's machine learning libraries, such as scikit-learn and TensorFlow, can be advantageous for this undertaking.
Python: Machine learning techniques can be employed to build a model that can classify sequels by examining a variety of various features and attributes. Python's machine learning libraries, such as scikit-learn and TensorFlow, offer significant benefits for performing this task.
Flask: Flask is a minimalistic web framework designed for Python. Flask can be utilized to develop a user-friendly online interface that facilitates interaction with machine learning models and enables access to database operations.
MySQL: MySQL is a robust relational database management system that enables the storage and management of several types of data associated with different projects, including training data, classification results, and user data.
Blockchain (Integrating for Scalability): Blockchain technology can be incorporated to enhance the scalability of data by ensuring its security and immutability, particularly when handling sensitive information. This effort deployed blockchain using truffle and ganache frameworks. Ethereum-specific development framework Truffle is popular. It gives developers complete tools for building, assessing, and executing intelligent contracts. Truffle simplifies blockchain development with a developer environment, testing system, and resource pipeline. Truffle easily integrates with Ganache, a development and testing Ethereum network. Ganache lets developers connect to simulated Ethereum accounts locally to test smart contract code.
5. Results and Discussion
The main aim of this work is, how can machine learning techniques be effectively applied to proteomics data to improve protein identification and quantification accuracy? To assess the effectiveness of the five classifiers, namely, random forest (RF), neural networks (NN), logistic regression (LR), support vector machines (SVM), and decision trees (DT), the performance measures employed include accuracy, precision, and recall. This section outlines three experiments that were undertaken to calculate the performance scores using two test sets. The test sets consist of 20% and 30% of the dataset, which will be used to evaluate the performance of the model.
Experiment 1:
This experiment was undertaken to determine the accuracy scores of the machine learning models utilizing two test sets and compare the results provided by Zhang and Team
| [8] | Singh and Tripathi, 2016. Singh, U. and Tripathi, S. (2016). Protein classification using hybrid feature selection technique. In International Conference on Smart Trends for Information Technology and Computer Communications, pages 813–821. Springer. ISBN: 978-981-10-3432-9. |
[8].
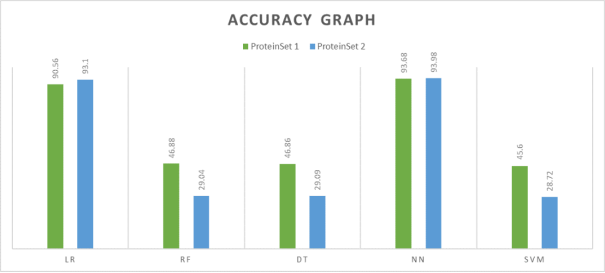
The accuracy results for all the classifiers are shown in
Table 2.
Figure 3 illustrates a comparative study of the accuracy of all five machine learning models.
Table 2. Accuracy results.
Classification Model | Protein testset 1 (%) | Protein testset 2 (%) |
LR | 90.56 | 93.1 |
RF | 46.88 | 29.04 |
DT | 46.86 | 29.09 |
NN | 93.68 | 93.98 |
SVM | 45.6 | 28.72 |
Table 2 demonstrates that the Neural Network classification technique outperforms other classification methods when evaluated on two different test sets. It is evident that the difference in accuracy measures between of the two test sets of the Neural Networks is negligible. The accuracy values of the other classifiers except logistic regression are significantly poor.
Figure 3. Accuracy comparison graph.
Experiment 2:
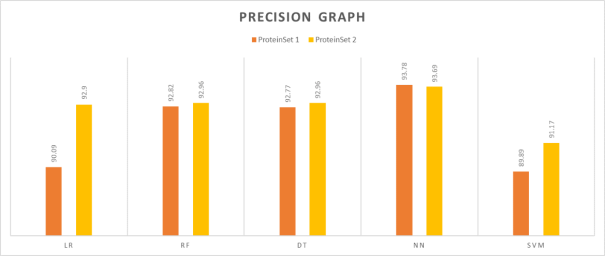
This experiment was conducted to determine the precision scores of the ML models with two test sets. The precision results for all the classifiers are shown in
Table 3.
Figure 4 illustrates a comparative study of the precision of all five machine learning models with performance with different deep-learning techniques described by Agarwal
and Zhang
| [9] | Zhang, Y., Li, X., & Wang, Y. (2023). Proteomics data analysis using machine learning on AWS. Bioinformatics, 40(10), 1839-1846. |
[9]
works.
Table 3 shown that the Neural Network algorithm outperforms other algorithms when evaluated on two different test sets. It is evident that the difference in precision measures between of the two test sets of the Neural Networks is negligible.
Table 3. Precision results.
Classification Model | Protein testset 1% | Protein testset 2% |
LR | 90.09 | 92.9 |
RF | 92.82 | 92.96 |
DT | 92.77 | 92.96 |
NN | 93.78 | 93.69 |
SVM | 89.89 | 91.17 |
Figure 4. Precision comparison graph.
Experiment 3:
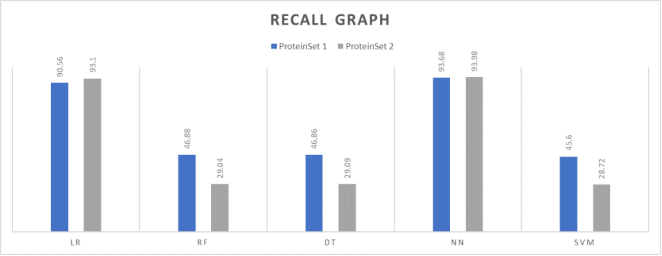
This experiment was conducted to determine the recall scores of the classification models with two test sets. The recall results for all the classifiers are shown in
Table 4.
Figure 5 illustrates a comparative study of the recall scores of all five machine learning models.
Table 3 shown that the Neural Network algorithm outperforms other algorithms when evaluated on two different test sets. It is evident that the difference in precision measures between of the two test sets of the Neural Networks is negligible. The recall values of the other classifiers except logistic regression are significantly poor.
Table 4. Recall results.
Classification Model | Protein testset 1% | Protein testset 2% |
LR | 90.56 | 93.1 |
RF | 46.88 | 29.04 |
DT | 46.86 | 29.09 |
NN | 93.68 | 93.98 |
SVM | 45.6 | 28.72 |
Figure 5. Recall comparison graph.
6. Conclusion and Future Work
Proteomics, the scientific discipline that investigates the properties and roles of proteins in living organisms, has experienced a significant rise in the amount of data it generates. This trend brings out several prospects and difficulties. This project aims to fulfil the requirement for sophisticated data analytics and data integrity in the field of proteomics research. By leveraging the capabilities of Machine Learning (ML), Blockchain technology, objective is to revolutionize proteomics research. There are five distinct classification algorithms used for protein sequence classification and predictions: Random Forest (RF), Logistic Regression (LR), Neural Networks (NN), Support Vector Machines (SVM), and Decision Trees (DT). Performed an experimental analysis for classification and predictions using two test sets, each consisting of 20% and 30% of the dataset, respectively. The experimental findings demonstrate the superior performance of the Neural Network algorithm compared to other algorithms in the classification task. This work successfully achieved Scalability, Availability, Data integrity, and security by incorporating blockchain technologies. Future research can plan to investigate more suitable approaches for protein sequence comparison and deep learning models to enhance performance and decrease computing costs.
Abbreviations
DNA: Deoxyribonucleic Acid
DT: Decision Trees
FN: False Negative
FP: False Positive
GAN: Generative Adversarial Network
LR: Logistic Regression
ML: Machine Learning
NLP: Natural Language Processing
NN: Neural Networks
PDB: Protein Data Bank
RCSB: Research Collaboratory for Structural Bioinformatics
RF: Random Forest
RNA: Ribonucleic Acid
SVM: Support Vector Machine
TF-IDF: Term Frequency-Inverse Document Frequency
TN: True Negative
TP: True Positive
Acknowledgments
I would like to confirm the article I am submitting is my original work and has no conflict of interest/plagiarism issues.
Author Contributions
Preethi Kolluru Ramanaiah is the sole author. The author read and approved the final manuscript.
Funding
This work is not supported by any external funding.
Conflicts of Interest
The author declares no conflicts of interest.
References
| [1] |
J. Bernardes and C. Pedreira, (2013), "A Review of Protein Function Prediction Under Machine Learning Perspective," Recent Patents on Biotechnology, vol. 7, no. 2, pp. 122–141.
http://dx.doi.org/10.2174/18722083113079990006
|
| [2] |
Aggarwal, Divyanshu & Hasija, Yasha. (2022). A Review of Deep Learning Techniques for Protein Function Prediction.
https://doi.org/10.48550/arXiv.2211.09705
|
| [3] |
Karunapala, 2015. Karunapala, E. (2015). Protein Function Prediction Using Machine Learning. PhD thesis.
|
| [4] |
Piovesan et al., 2015. Piovesan, D., Giollo, M., Leonardi, E., Ferrari, C., and Tosatto, S. C. (2015). Inga: protein function prediction combining interaction networks, domain assignments and sequence similarity. Nucleic acids research, 43(W1): W134–W140.
http://dx.doi.org/10.1093/nar/gkv523
|
| [5] |
Kotlyar et al., 2014. Kotlyar, M., Pastrello, C., Pivetta, F., Sardo, A. L., Cumbaa, C., Li, H., Naranian, T., Niu, Y., Ding, Z., Vafaee, F., et al. (2014). In silico prediction of physical protein interactions and characterization of interactome orphans. Nature methods, 12(1): 79
https://doi.org/10.1038/nmeth.3178
|
| [6] |
Rentzsch and Orengo, 2013. Rentzsch, R. and Orengo, C. A. (2013). Protein function prediction using domain families. In BMC bioinformatics, volume 14, page S5. BioMed Central.
https://doi.org/10.1186/1471-2105-14-S3-S5
|
| [7] |
Z. He, S. Zhang, F. Gu, and J. Wu, (2019). Mining conditional discriminative sequential patterns, Inf. Sci., vol. 478, pp. 524–539.
http://dx.doi.org/10.1016/j.ins.2018.11.043
|
| [8] |
Singh and Tripathi, 2016. Singh, U. and Tripathi, S. (2016). Protein classification using hybrid feature selection technique. In International Conference on Smart Trends for Information Technology and Computer Communications, pages 813–821. Springer. ISBN: 978-981-10-3432-9.
|
| [9] |
Zhang, Y., Li, X., & Wang, Y. (2023). Proteomics data analysis using machine learning on AWS. Bioinformatics, 40(10), 1839-1846.
|
| [10] |
Goodfellow I.; Pouget-Abadie J.; Mirza M.; Xu B.; Warde-Farley D.; Ozair S.; Courville A.; Bengio Y. (2020). Generative adversarial networks. Communications of the ACM 2020, 63 (11), 139–144.
https://doi.org/10.1145/3422622
|
| [11] |
Z. He, G. Xu, C. Sheng, B. Xu and Q. Zou, "Reference-Based Sequence Classification," in IEEE Access, vol. 8, pp. 218199-218214, 2020,
https://doi.org/10.1109/ACCESS.2020.3042757
|
| [12] |
Agarwal, Ankita & Singh, Kunal & Kaushik, Shri Kant & Bahadur, Ranjit. (2022). A comparative analysis of machine learning classifiers for predicting protein-binding nucleotides in RNA sequences. Computational and Structural Biotechnology Journal. 20.
https://doi.org/10.1016/j.csbj.2022.06.036
|
| [13] |
Structural Protein Sequences. (2018). Kaggle. Source:
https://www.kaggle.com/datasets/shahir/protein-data-set
last accessed 2023/11/15.
|
| [14] |
Research Collaboratory for Structural Bioinformatics. Source:
https://www.rcsb.org/
last accessed 2023/11/15
|
| [15] |
Jiang, Mudi & Wang, Jiaqi & Hu, Lianyu & He, Zengyou. (2023). Random forest clustering for discrete sequences. Pattern Recognition Letters. 174. 10.1016/j.patrec.2023.09.001.
http://dx.doi.org/10.1016/j.patcog.2024.110388
|
| [16] |
Chaudhary, M. (2021). TF-IDF Vectorizer scikit-learn - Mukesh Chaudhary - Medium. Medium. Source:
https://medium.com/@cmukesh8688/tf-idf-vectorizer-scikit-learn-dbc0244a911a
last accessed 2023/11/05.
|
| [17] |
Preethi Kolluru (2023) Breast Cancer Classification Using Transfer Learning with Ensemble
https://doi.org/10.15680/IJIRCCE.2024.1202006
|
| [18] |
Sklearn. ensemble. Random Forest Classifier. (n.d.). Scikit-learn. Source:
https://scikit-learn.org/stable/modules/generated/sklearn.ensemble.RandomForestClassifier.html
last accessed 2023/11/01
|
| [19] |
What is Logistic Regression? - Logistic Regression Model Explained - AWS. (n.d.). Amazon Web Services, Inc. Source:
https://aws.amazon.com/what-is/logistic-regression/
last accessed 2023/11/01
|
| [20] |
Liang and Bose, 1996. Liang, P. and Bose, N. (1996). Neural network fundamentals with graphs, algorithms, and applications. McGraw-Hiil, New York.
|
| [21] |
Support Vector Machines. (n.d.). Scikit-learn. Source:
https://scikit-learn.org/stable/modules/svm.html
last accessed 2023/11/01
|
Cite This Article
-
-
@article{10.11648/j.ajai.20240801.13,
author = {Preethi Kolluru Ramanaiah},
title = {Proteomics Data Classification Using Advanced Machine Learning Algorithm
},
journal = {American Journal of Artificial Intelligence},
volume = {8},
number = {1},
pages = {13-21},
doi = {10.11648/j.ajai.20240801.13},
url = {https://doi.org/10.11648/j.ajai.20240801.13},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ajai.20240801.13},
abstract = {Proteomics, the study of proteins and their functions within biological systems, has become increasingly data-intensive, presenting both opportunities and challenges. This project addresses the need for advanced data analytics and data integrity in proteomics research. Leveraging the power of machine learning (ML) and blockchain technology, this attempt aims to transform proteomics research. This work encompasses three key objectives. First, collect, clean, and integrate proteomics data from diverse sources, ensuring data quality and consistency. Second, employ ML algorithms to analyze this data, revealing crucial insights, identifying proteins, and predicting their functions. Third, implement blockchain technology to safeguard the authenticity and integrity of the proteomics data, providing an auditable and tamper-proof record. Implemented a user-friendly web interface, facilitating collaboration among researchers and scientists by granting access to shared data and results. This study included various classification methods for the investigation of protein classification, namely, random forests, logistic regression, neural networks, support vector machines, and decision trees. In conclusion, the proposed work is poised to revolutionize proteomics research by enhancing data analytics capabilities and securing data integrity, thereby enabling scientists to make more informed and confident discoveries in this critical field.
},
year = {2024}
}
 Copy
|
Copy
|
 Download
Download
-
TY - JOUR
T1 - Proteomics Data Classification Using Advanced Machine Learning Algorithm
AU - Preethi Kolluru Ramanaiah
Y1 - 2024/05/17
PY - 2024
N1 - https://doi.org/10.11648/j.ajai.20240801.13
DO - 10.11648/j.ajai.20240801.13
T2 - American Journal of Artificial Intelligence
JF - American Journal of Artificial Intelligence
JO - American Journal of Artificial Intelligence
SP - 13
EP - 21
PB - Science Publishing Group
SN - 2639-9733
UR - https://doi.org/10.11648/j.ajai.20240801.13
AB - Proteomics, the study of proteins and their functions within biological systems, has become increasingly data-intensive, presenting both opportunities and challenges. This project addresses the need for advanced data analytics and data integrity in proteomics research. Leveraging the power of machine learning (ML) and blockchain technology, this attempt aims to transform proteomics research. This work encompasses three key objectives. First, collect, clean, and integrate proteomics data from diverse sources, ensuring data quality and consistency. Second, employ ML algorithms to analyze this data, revealing crucial insights, identifying proteins, and predicting their functions. Third, implement blockchain technology to safeguard the authenticity and integrity of the proteomics data, providing an auditable and tamper-proof record. Implemented a user-friendly web interface, facilitating collaboration among researchers and scientists by granting access to shared data and results. This study included various classification methods for the investigation of protein classification, namely, random forests, logistic regression, neural networks, support vector machines, and decision trees. In conclusion, the proposed work is poised to revolutionize proteomics research by enhancing data analytics capabilities and securing data integrity, thereby enabling scientists to make more informed and confident discoveries in this critical field.
VL - 8
IS - 1
ER -
Copy
|
Download